6. Appendix¶
6.1. Pre-processing miRBase data:¶
The MIRfix pipeline [21] provides the general core of
functions to curate bona fide metazoan microRNA annotations. To make use of
this curation process, it is fundamental to organize the input data in a
specific format, as referenced in more detail in [21]. In
summary, it is required:
A set of precursor sequences with their associated mature sequences.
Genome sequences from which miRNAs were annotated.
A relation file that describes the relation between precursors and their annotated matures.
Additional parameters are required, but they did not depend from external information/databases.
On miRNAture the source of the curation data has been obtained from a re-evaluation
of the annotations deposited on miRBase v.22.1 [7]. In this
version, miRBase accounted for a set of X canonical and non-canonical miRNA families,
from which Y are constituted by metazoan sequences. Internally, miRNAture
performs an evaluation of the canonical model, that relies on the correct
positioning cleavages performed by Drosha and Dicer at the precursor maturation steps.
Computationally, this is translated on a correct position of the mature sequence,
accurate delimitation of precursor, and a phylogenetic support, addressed by the
construction of family mature-anchored structural alignments. As previously
reported for the Rfam miRNA families [19], an iterative
assessment involves a selection of sequences, consistent criteria to evaluate
the miRNAs and their mature products, and generation of probabilistic models
derived from anchored-alignments to search additional candidates that would
incorporate defined curation criteria. This criteria was inherited to perform an
evaluation of the miRBase metazoan families and generate the corrected
dataset that miRNAture uses to evaluate new candidates and their maturation
entities.
As a toy example, the family miR-17 (MIPF0000001) was selected to demonstrate
the assessment steps performed over all pre-calculated dataset used by miRNAture.
As reported in miRBase this family is composed by 239 miRNA precursors derived from
39 vertebrate species. Through the filtering approach the following subsetting steps are
considered:
Remove non-metazoan sequences.
Filter duplicates (which share 100% identity) and select one representant sequence.
In this family, 117 duplicated sequences where recognized. For instance the sequence bta-mir-18a (MI0004740) from Bos taurus has shown 21 orthologs, as follows:
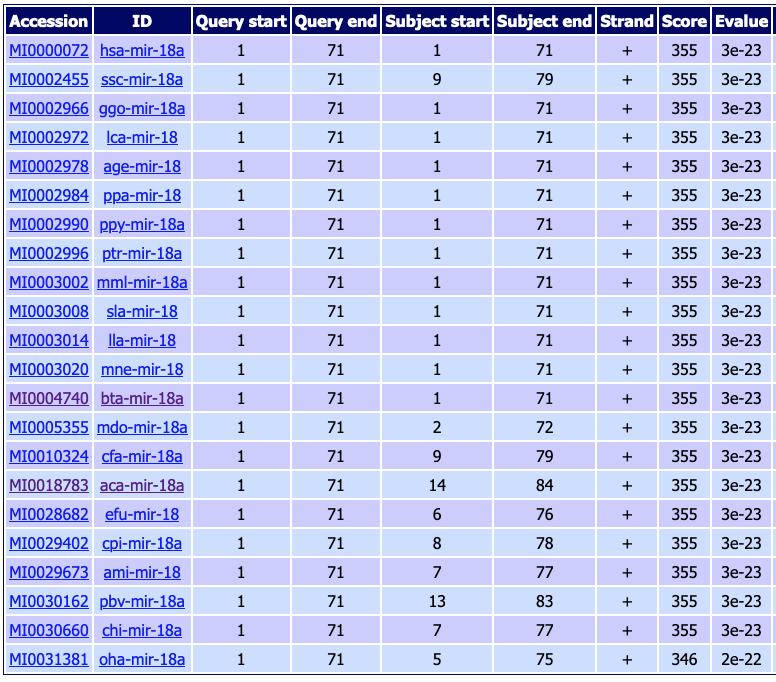
Fig. 6.1 Identified orthologs from bta-mir-18a on vertebrates.¶
And some corresponding alignments:
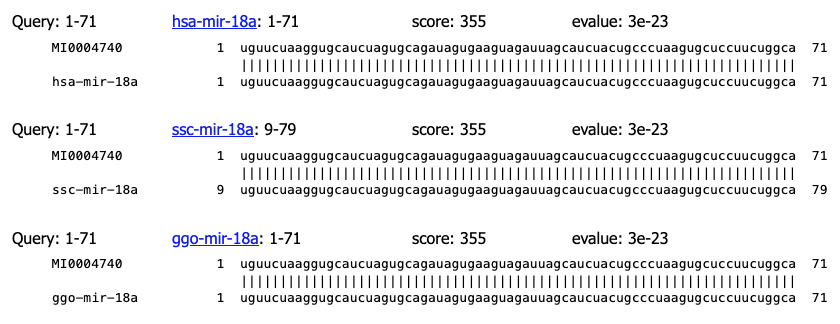
Fig. 6.2 Alignments as evidence of 100% identity.¶
Remaining 122 families were subject of a structural assessment by MIRfix,
which filtered 4 sequences based on the incorrect miRNA folding in regard their
annotated mature sequences, and one sequence contained a bad positioned mature
sequence in the reported precursor, a successful extension of the precursor based
on the miR and miR* prediction, rescued the candidate.
Category |
Accession numbers |
|---|---|
Bad position mature sequences |
MI0004822 |
Filtered sequences |
MI0012797, MI0012947, MI0019542, and MI0013837 |
At the end of the assessment 118 sequences passed all filters to be considered
into the curation dataset used on miRNAture.
The same approach curated all metazoan miRNA families from miRBase (1415),
validating about 79% (1111) of the families and setting the curation dataset used on
miRNAture.