Proteinortho - Orthology detection tool
Description
This program finds orthologous proteins within different species.
It can either be started giving the intended files in fasta-format (at least
two) after the OPTIONS or just one file containing the paths to these. This
is especially useful if their number grows. Each file should represent all
proteins (or the part of it that should be investigated) of one species.
In a first step all files are blasted against each other. The hits will
be evaluated according to the given OPTIONS and transformed into a graph,
where each protein is represented by a node. This graph will be fragmented
into its connected components, thus proteins which are connected to each
other.
Proteinortho was designed to deal with large data sets and also behave
nicely regarding the memory consumption. However, it works for small sets as well.
Details about the algorithm and benchmarks can be found in this thesis.
- Important:
- Protein ids must be globally different! You should also consider
that blast may cut the ids on a whitespace using the first part only.
Output Format
The OUTPUT is a tab separated matrix.
First line starts with # followed by the file names. Second line starts
with # followed by the corresponding number of proteins in the files.
From here each line represents a connected component and therefore the
ids of determined orthologous proteins.
Syntax
proteinortho.pl
[OPTIONS] <FILES> >OUTPUT
proteinortho.pl [OPTIONS] <FILE> >OUTPUT
Options
- -e=E-VALUE
- E-VALUE for blasts
[default: 1e-10]
- -a=#THREADS
- number of THREADS to make use of dual- and multi-core
CPUs
[default: 1]
- -p=blastp|blastn
- defines the blast program
[default: blastp]
- -r=0|1
- enables or disables reciprocal the blast condition
[default: 1 (enabled)]
- -m=(0..1)
- minimum similarity of best blast hits
allowed are doubles within the interval (0..1)
all hits with
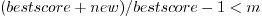 are included
are included
takes all hits into account
1 only the best (maybe more with equal bitscore)
useful to handle paralogous better
[default: 0.95 (nearly equal)]
- -selfblast
- applies an additional blast for every species against itself
this may increase the detection of paralogs, but is normally not necessary a similar hits are found if -m is not set to 1
- -f
- force blastall (even if blast output is found)
- -ff
- force
formatdb (even if databases are found)
- -remove
- removes blast outputs after
use
- -verbose
- gives information about what happens, including a progress report
and a lasting time approximation
- -dir=DIRECTORY
- defines the DIRECTORY
for the blast outputs
[default directory: working directory]
- -cmat
- includes putative paralogous proteins to the output
sets which contain such proteins are not reported otherwise
paralogous protein ids are separated by ","
[default file: cc.matrix]
- -debug
- keeps temporary files for debugging
- -plog[=FILE]
- logfile for pairwise blast hits
[default file: pb.log]
- -plog[=FILE]
- logfile for connected components
[default file: cc.log]
- -ulog[=FILE]
- ultimate logfile, this is actually a
post-process of plog and clog
the creation is very time intensive and not recommended if more than 10,000
proteins are involved
[default file: ultimate.log]
Multiple Machine Options
The main part of Proteinortho
consists of blasting each species against each other. This can take several
hours up to days if hundreds of species are involved - even on multi-core
machines. For this purpose a mechanism has been implemented which allows
to distribute that workload over multiple machines.
Every option aside from -a=#THREADS needs to be the same. This is especially
important for the directory in which the blasts are stored. A file named
sync will be created their and used to synchronize the processes. As flock
is not capable for network file systems a temporary directory named lock/
is used for locking. Both may need to be removed if Proteinortho was interrupted
or crashed and a restart is intended.
Run all scripts using the option
- -blastonly
As the scripts synchronize themselves the order or time you start it on
different machines does not matter. You can even stop certain processes
if needed. See SIGNALS for more details to that topic. After the blasts are
done, all started scripts will be terminated.
If that happened, you can grab the results and finish the calculations.
Start the script again on one machine using the same options as before.
Instead of -blastonly use the option
- -blastdone
-
Examples
To run this program the standard way comparing
two or more species type:
proteinortho.pl speciesA.faa speciesB.faa >orthologs.out
If you want to define the number of threads, have live progress report
and store blast files in a separate folder, type:
mkdir blastout
proteinortho.pl
-a=4 -verbose -dir=blastout/ files.list >orthologs.out
Copyright
Copyright ©2009
Free Software Foundation, Inc. License GPLv2+: GNU GPL version 2 or later
http://gnu.org/licenses/gpl.html
This is free software: you are free to change and redistribute it.
There is NO WARRANTY, to the extent permitted by law.
Reporting Bugs
Marcus
Lechner marcus[at]bioinf[dot]uni-leipzig[dot]de
Authors
Written by Marcus Lechner, Lydia
Steiner and Sonja J. Prohaska
Bioinformatics Group, Department of Computer Science, and Interdisciplinary Center for Bioinformatics, University of Leipzig
See also
orthomatrix2tree.pl a tool which allows to generate trees based on the shared proteins