|
Home
CV
Research
Publications
Links
Animations
|
Research
Simulation of metabolic networks
The origin and evolution of metabolism is an interesting
field of research with many unsolved
questions. Simulation approaches have proven
to be helpful in explaining properties observed
in real world metabolic reaction networks.
We propose here a more complex and intuitive
graph-based model combined with an
artificial chemistry. Instead of differential
equations, enzymes are represented as graph
rewriting rules and reaction rates are derived
from energy calculations of the involved
metabolite graphs.
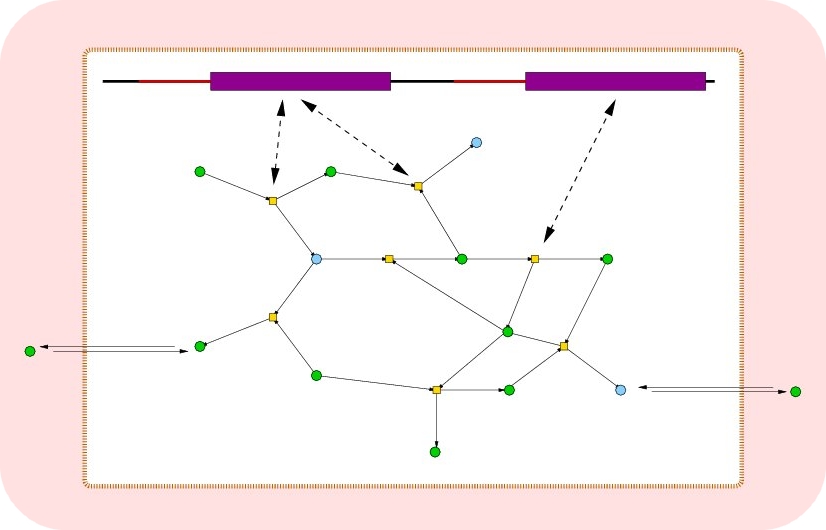
Initially, the environment (set of metabolites)
and the chemistry (set of chemical reactions)
are defined and a population of cells containing a
genome and a (empty) metabolism are added
to the simulation. The genes are expressed
by mapping their RNA-sequence to an enzyme
function. Enzymes look for reactants, produce
new metabolites and thus build up the
metabolic reaction network.
In each generation, individuals are selected
based on their metabolic yield and then create
new individuals with mutated genomes.
Analysis of metabolic networks
Metabolic pathway analysis is the calculation and analysis of the pathway distribution of a steadystate
metabolic network to gain insights about its structure, functionality and properties. The calculation
starts with the formation of the stoichiometric matrix presentation of the network and delivers
the extreme pathways, spanning the entire steady state flux space, as the final result.
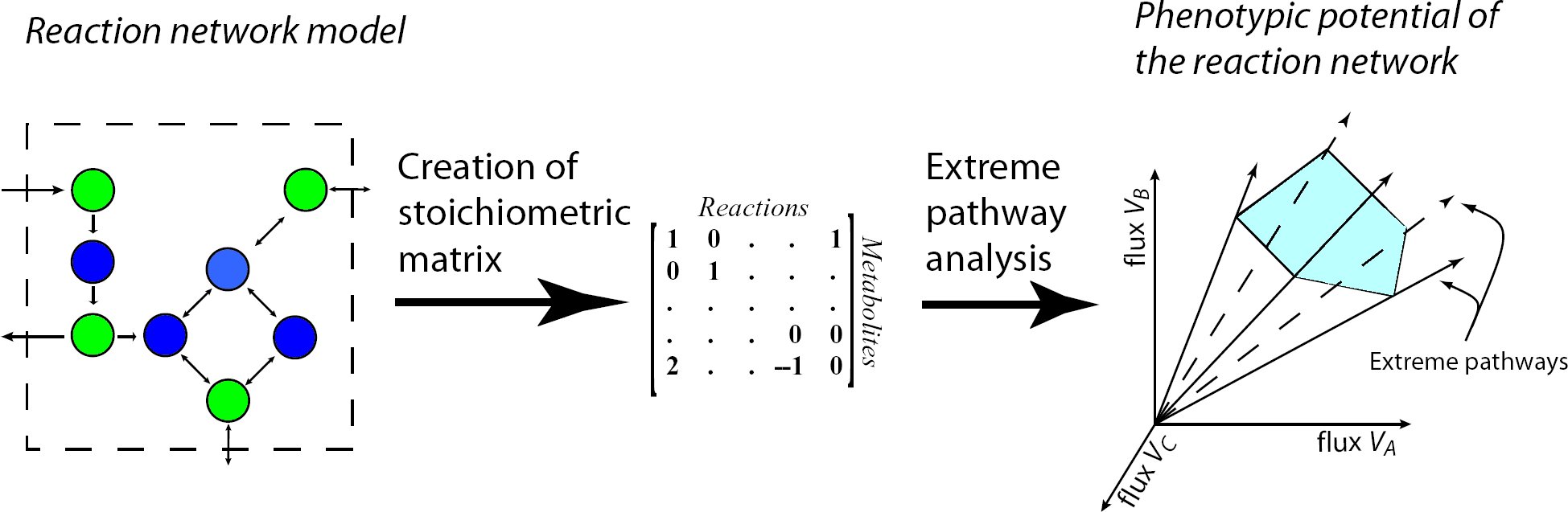
Extreme Pathways are the set of essential pathways through which all other possible pathways of
the metabolic network can be generated, they are also minimal in the sense that they do not consist
of smaller pathways.
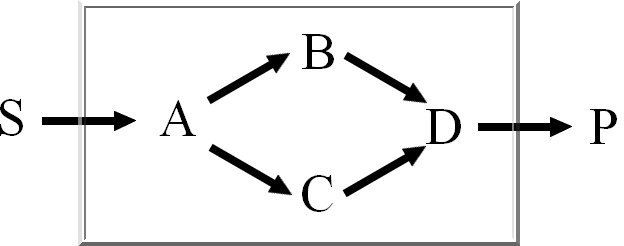 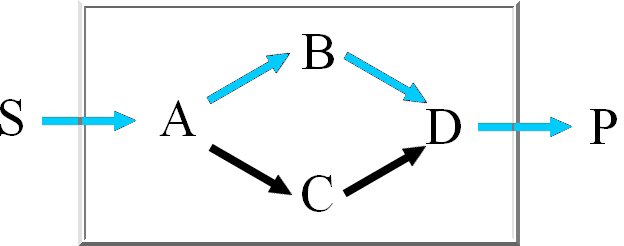 
We calculate the set of extreme pathways with an improved binary Null-space approach. The computation
time of the existing approach is dependent on the row-ordering. We integrated a new way
to order the rows to reduce the number of candidate pathways.
Minimal knockout sets are sets of reactions that need to be removed in order to disable the function
of a certain target reaction, this means that there may not be any extreme pathway containing this
target reaction.
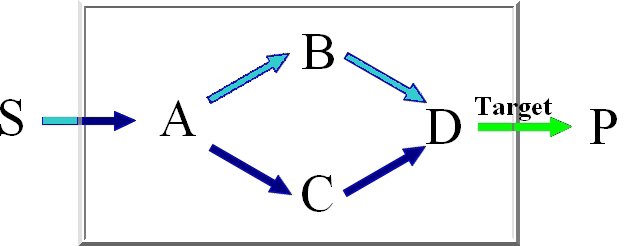 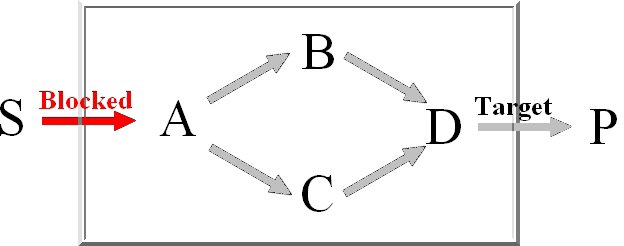 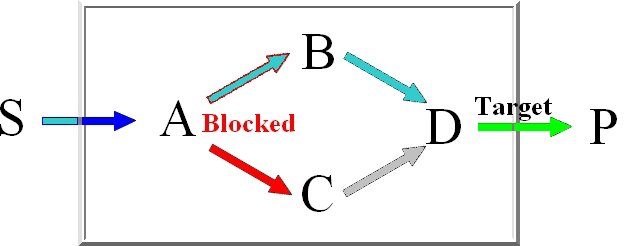 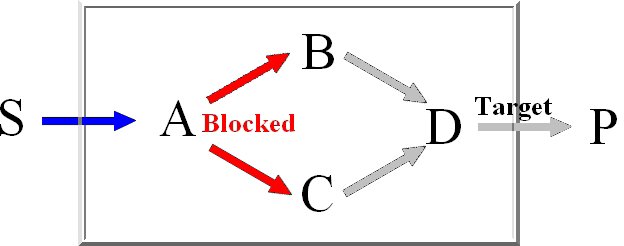
For the computation of the minimum knockout sets we implemented an improved depth-first
version of Berge's algorithm which avoids the costly superset removal.
Emergence of network properties
The emergence and evolution of properties on the network level, is another
intriguing question. Here we apply our metabolic pathway analysis
tool to our simulation for the evolution
of metabolisms. In this process the tool is used to define a realistic selection
criteria -optimal metabolic yield- for the
metabolisms, leading to networks with properties
as observed in their real-world counterparts
(e.g. scale free degree distribution). Further we use it to
analyze less well understood properties of the
resulting networks, such as robustness or modularity,
to make predictions about their origin
and development. It can also be used as a standalone
tool for the enumeration of elementary
modes and the subsequent computation of minimal
cut-sets, in a very memory efficient way.
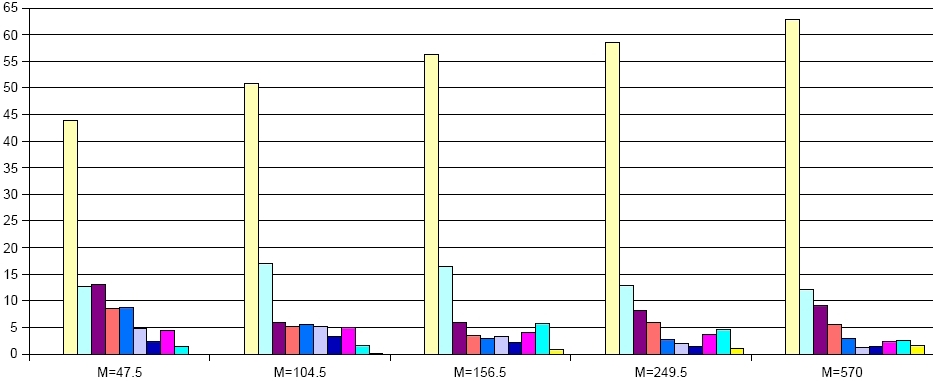
Robustness of a network can be defined as a property of the underlying graph structure or as a dynamical property of the system that is represented by the network.
Simple graph-based measures for robustness are for example, degree or connectivity distributions, as well as the entropy. Measures for the robustness of the system dynamics, especially in metabolic networks, are related to fluxes through the system and their behavior to eleimination of edges (knockout of genes/enzymes) and nodes (depletion of metabolites). Here we use several measures based on the set of extreme pathways and the size distribution of the above described minimal knockout sets.
Genotype-Phenotype mappings
For many years it is known that neutral mutations have a considerable influence on the evolution in
molecular systems. The folding of RNA sequences to secondary structures with its many-to-one property
represents a mapping entailing considerable redundancy. Various extensive studies concerning RNA folding
in the context of neutral theory yielded insights about properties of the structure space and the mapping
itself. We intend to get a better understanding of some of these properties and especially of the evolution of
RNA-molecules as well as their effect on the evolution of the entire molecular system.
We introduce a novel genotype-phenotype mapping based on the relation between RNA sequence and its
secondary structure for the use in evolutionary studies. The inspiration for this particular mapping emerged
from the modeling of RNA enzymes within our simulation framework for the evolution of metabolic reaction
networks. The genome contains a number of RNA genes which then give rise to RNA enzymes acting on metabolites and
thus shaping the metabolic network. The use of our mapping allows not only for a more realistic
study of the evolution of the entire system, but also enables us to observe the behavior of our enzymes itself
and therefore possibly gain some insights about the evolution of catalytic molecules in general.
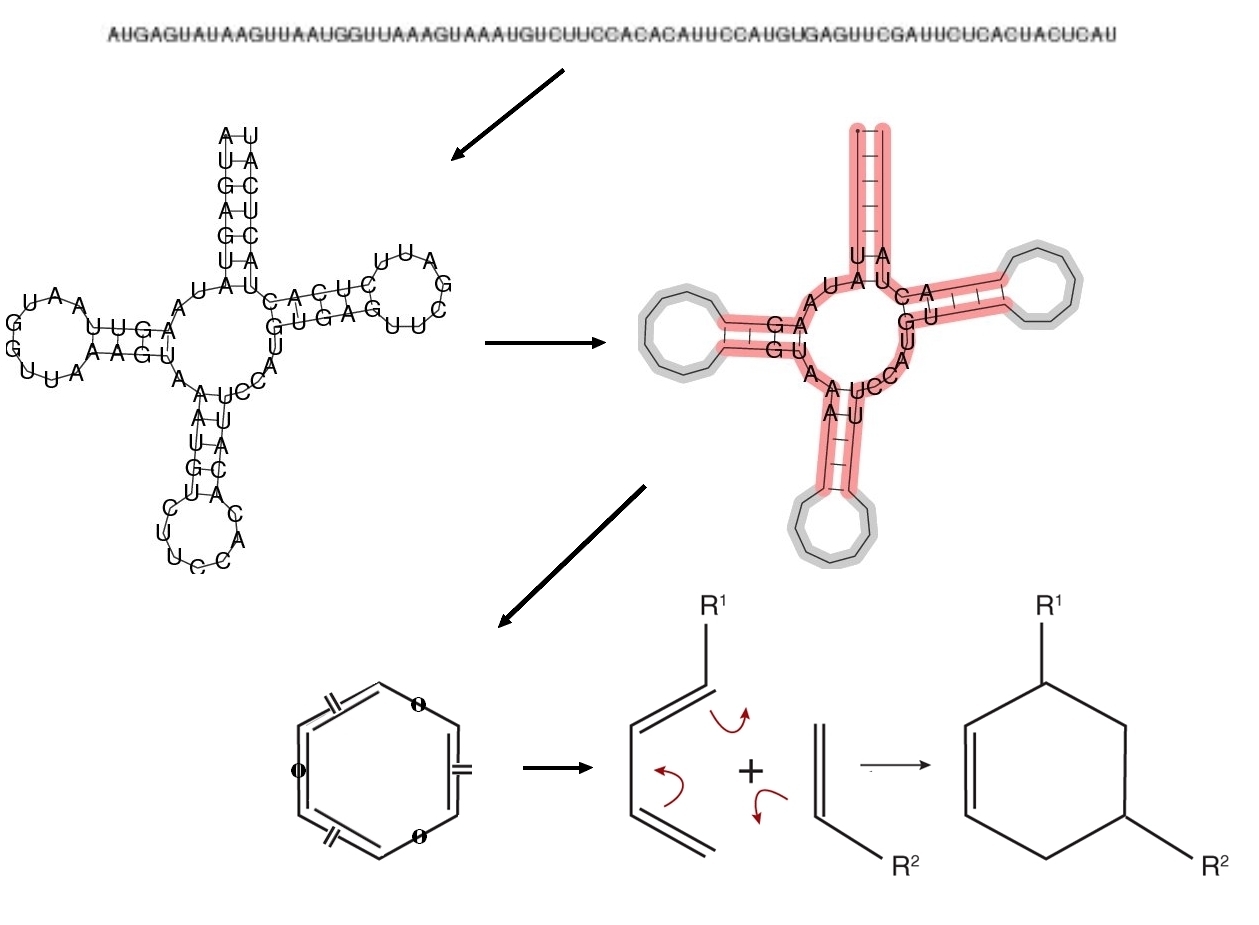
Enzymes typically have an active site where only few amino acids or bases determine its catalytic function
and the remaining structure has mostly stabilization function. Accordingly, we extract structural and sequence
information only from a restricted part of the fold. We decided to focus on the longest loop of the folded
RNA. The idea for mapping the extracted information to a specific chemical reaction was encouraged by
the fact that many enzymes catalyze a reaction by stabilizing its transition state. Recent work on hairpin
ribozymes and other catalytic RNA support that as a common strategy for RNA enzymes. Given the definition
of Fujita's imaginary transition structures (ITS), we developed a unique index for all possible pericyclic
chemical reactions, describing the constitution of the reaction's transition state. Every RNA molecule is
assigned such an reaction ID based on the information from its fold. The length of the longest loop specifies
the number of involved atoms and the sequence within the loop determines the atom types. The bond types
are derived from structural characteristics of the loop, such as the length and position of contained stems.
Thus, a mapping from RNA sequence (genotype) to a chemical reaction (phenotype) is produced.
Besides using the mapping in several simulation runs which yielded realistic metabolic networks and
connectivities, we performed several statistical tests commonly used in neutral theory, such as the number
of visited phenotypes and the average discovery rate during a random neutral walk. We compared it with
results of approaches using cellular automatons, random boolean networks and other mappings based on
RNA folding. It exceeds all non-RNA mappings in extent and connectivity of the underlying neutral network.
Further, it has a significantly higher evolvability and innovation rate than the rest. Especially interesting is
the highly innovative starting phase in RNA-based mappings.
Protein function prediction
Predicting Function of proteins is an important problem in computational
biology and many approaches have been introduced to solve this problem.
However, there is no single approach which regards all issues of protein function
prediction. The most promising clue to infer function seems to be Protein
Packing Motifs, patterns of only a few amino acids which are spatially close
together and possess similar bio-chemical and geometrical properties. Our
approach finds these packing motifs by mining closed frequent subgraphs
from a set of protein graphs. The key to finding good motifs is the use of
optimal labels for nodes and edges of the protein graphs, thus we introduce a
novel way to assign these labels based on credible biological characteristics
of such motifs. The use of Feature Selection allows the approach to be extended
with discriminative features other than our motifs. Possible additional
features could come from other function prediction approaches. By assigning
feature vectors to protein graphs we have the possibility to use our approach
to develop a protein index for structural protein databases, in particular the
PDB databse, since we extract all information for our approach solely from
this database.
A packing motif is a pattern, representing a cluster of residues which are close in
space with no restriction in sequence, e.g. distance or order, which occurs multiple
times in a set of proteins. Due to that definition they can hardly be found by
sequence analysis, thus our approach uses structural information.
It is known that protein packing motifs can provide strong evidence for bio-chemical
function, for example the function of the HIV virus was identified through a characteristic
motif of this functional family.
Active sites of enzymes, which are also packing patterns, have usually different
bio-chemical properties than the all other areas on the enzyme surface. For example
ligand binding sites can be distinguished from other parts due to a high number
of hydrophobic amino acids, which are mostly in the core of the protein rather
than the surface. Other characteristics which can be used to detect such areas, are
clusters of non-polar amino acids or certain combinations of polar amino acids.
Considering this we decided to use these attributes to form the labels for the nodes
in the protein graphs. We discarded the idea of using the amino acid type, because
conservation in a packing motif means rather of a class than of a specific type.
Response network analysis
One important aspect of computational systems biology includes the
identification and analysis of functional response networks within
large biochemical networks. These functional response networks
represent the response of a biological system under a particular
experimental condition which can be used to pinpoint critical
biological processes.
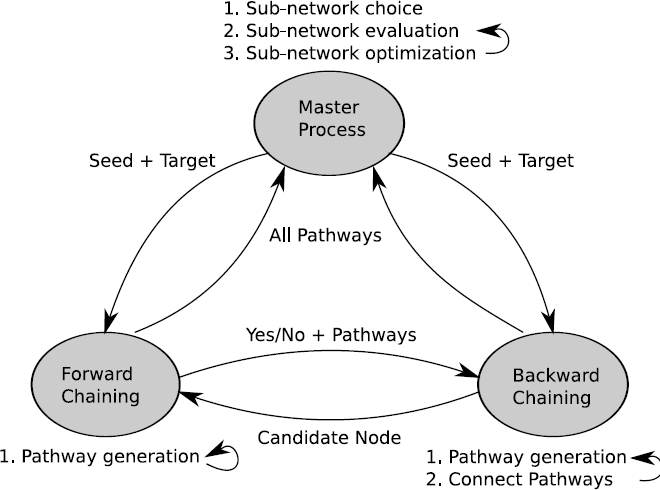
For this purpose, we have developed a novel algorithm to calculate
response networks as scored/weighted sub-graphs spanned by k-shortest
simple (loop free) paths. The k-shortest simple path algorithm is
based on a forward/backward chaining approach synchronized between
pairs of processors. The algorithm
scales linear with the number of processors used. The algorithm
implementation is using a Linux cluster platform, MPI lam and mpiJava
messaging as well as the Java language for the application.
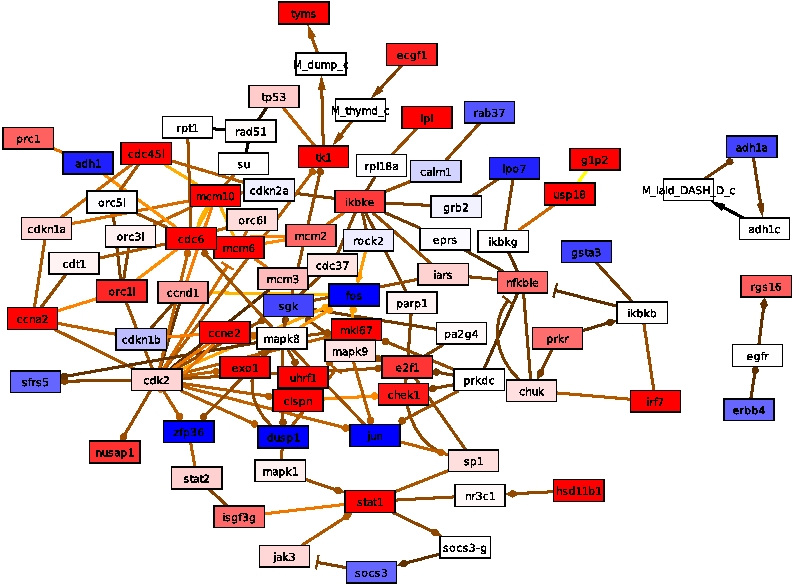
The algorithm is performed on a hybrid human network consisting of 45,041
nodes and 438,567 interactions together with gene expression
information obtained from human cell-lines infected by influenza
virus. Its response networks show the early innate immune response
and virus triggered processes within human epithelial cells. Especially
under the imminent threat of a pandemic caused by novel influenza strains,
such as the current H1N1 strain, these analyses are crucial for a
comprehensive understanding of molecular processes during early phases
of infection. Such a systems level understanding may aid in the
identification of therapeutic markers and in drug development for
diagnosis and finally prevention of a potentially dangerous disease.
|