blockbuster

detect blocks of overlapping reads using a gaussian-distribution approach
Langenberger D, Bermudez-Santana C, Hertel J, Hoffmann S, Khaitovitch P, Stadler PF
1. Introduction
Once short read sequences are mapped to a reference genome, one will face the problem of dividing consecutive reads into blocks to detect specific expression patterns. Due to biological variability and sequencing inaccuracies, the read arrangement does not always show exact block boundaries. The blockbuster tool automatically assigns reads to blocks and gives a unique chance to actually see the different origins where the short reads come from.
-
2.Method
In the first step, a mapped read u with start and end positions au and bu is replaced by a Gaussian density ρu with meanμu = (bu + au )/2 and variance σu2 . We set σu = s|(bu − au )/2|, where s is a parameter that is used to tune the resolution. For each locus, these Gaussian densities are added up separately for the two reading directions. The resulting curves f + and f − that exhibit pronounced but smooth peaks centered at blocks of reads with nearly identical midpoints (see Figure).
Now we use a greedy procedure to extract the reads that belong to the same block:
-
(1)Determine the location xˆ of the highest peak. Set B=∅ and δ=0.
-
(2)Include in the block B all reads u such that xˆ ∈[μu −(σu +δ),μu +(σu +δ)]. Set δ to the standard deviation of means μu, u∈B, and repeat step (2)
-
(3)ComputefB =u∈B ρu , output B, remove thereads in B, and set f → f −fB.
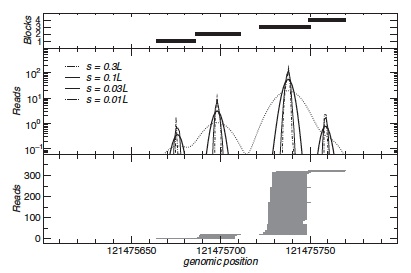
This procedure iteratively extracts blocks in an order that intuitively corresponds to their importance (see Figure). Since the area under a peak equals the number of reads in the block the height of the peaks provides a meaningful trade-off between the coherence of a block and its expression level. We therefore suggest to use the height of the peak to define the stop condition for blockbuster.
-
3. Quick Start
To build blockbuster from source you simply download the archive and extract it with
tar -xvf blockbuster.tar.gz
subsequently go to the new directory and type
make
First you have to map your sequenced tags to a reference genome (the easiest way to do that is to use segemehl) and safe the resulting read locations in a file using the bed format. The number of times each tag was sequences has be written in the score column of the file (tip: you should normalize this value by the number of times you can map this tag to the genome). The bed file has to be sorted by chromosome, strand, start pos and end pos of each tag.
Sort bed file:
sort -k 1,1 -k 6,6 -k 2n,2 -k 3n,3 -o sorted.bed unsorted.bed
Then you simply start blockbuster by typing
./blockbuster.x file.bed
In the archive you will also find a file called test.bed. This file contains human reads and it will show you how your input data for blockbuster has to look like.
There are two different output formats of blockbuster:
- block-ouput:
./blockbuster.x -print 1 test.bed (default)
OUTPUT:
>clusterID chrom clusterStart clusterEnd strand ClusterExpression tagCount blockCount
blockNb blockChrom blockStart blockEnd blockStrand blockExpression readCount
- tag-output:
./blockbuster.x -print 2 test.bed
OUTPUT:
>clusterID chrom clusterStart clusterEnd strand ClusterExpression tagCount blockCount
tagChrom tagStart tagEnd tagID tagExpression tagStrand blockNb
If you use segemehl for mapping, you can directly use the sorted segemehl output-file like this:
1. sort output file:
sort -k 10,10 -k 13,13 -k 11n,11 -k 12n,12 -o segemehl.sorted segemehl.out
2. run blockbuster:
./blockbuster.x -format 2 segemehl.sorted
-
4.Usage
blockbuster.x [OPTIONS] <file>
[OPTIONS]
-format <int>
-distance <int>
-minClusterHeight <float>
-minBlockHeight <float>
-scale <int>
-merge <int>
-tagFilter <int>
-print <int>
file format of input file (default: 1)
(1) bed
(2) segemehl-output
minimum distance between two clusters (default: 30)
minimum height (readno) of a cluster (default: 10)
minimum height (readno) of a block (default: 2)
scale stddev for a single read (default: 0.4)
merge reads with almost similar means (default: 0)
skip tags with expression smaller than this value (default: 0)
print out: (1) blocks (2) reads (default: 1)
5. Download
6. Contact
If you have any further questions, complaints or bug reports please mail to david@bioinf.uni-leipzig.de
7. Reference
If you use this program in your work you might want to cite:
Langenberger D, Bermudez-Santana C, Hertel J, Hoffmann S, Khaitovitch P, Stadler PF: "Evidence for Human microRNA-Offset RNAs in Small RNA Sequencing Data", Bioinformatics (2009) vol. 25 (18) pp. 2298-301