This page describes ADPfusion ≥ 0.1.0.0. The paper (local copy) describes how the backend (which the user shouldn’t need to see or know about) and one frontend based on the idea of “combinators” (symbolic functions between terminals and non-terminals).
In time for ICFP’12 a new version of ADPfusion ( ≥ 0.1) was ready the removes the need for combinators (except one) and greatly simplifies writing grammars.
ADPfusion is fast! It is possible to achieve performance very close to optimized C-code.
Contents
- Introduction to ADPfusion
- Runtime, vs. C and legacy ADP
- submitted paper
- source for the original submission
Introduction to ADPfusion: efficient, high-level dynamic programming in Haskell
Nussinov78: a simple (one table) dynamic programming algorithm
Let’s write the Nussinov ’78 algorithm using ADPfusion. First, we need (we don’t really need one but giving types is always a good idea) a signature. A signature collects the type of all functions required to evaluate a parse. We have 5 functions giving scores to parses (a. - e.) and a final function (f.) selecting the optimal parse. Function (d.), for example, builds RNA nucleotide pairs that bracket a subparses.
type Signature m a r =
( () -> a -- a.
, Char -> a -> a -- b.
, a -> Char -> a -- c.
, Char -> a -> Char -> a -- d.
, a -> a -> a -- e.
, SM.Stream m a -> m r -- f.
)Now that we have a signature, we need a grammar. The grammar defines the search space over the input. There is a single non-terminal “s”, and two terminals “b” for parsing a single character (or nucleotide) and the empty terminal parser “e”. The functions with our signature from above are (empty, up to h). We don’t specify them here as we want to use the same grammar with different evaluation functions.
We need to “INLINE” a lot to get optimal performance, the rule is to inline all outermost functions like “gNussinov”.
“transtoN” makes sure that the non-terminal “s” in “split” is a variant that parses only non-empty (hence N) subtrees. Otherwise “split” diverges.
gNussinov (empty,left,right,pair,split,h) s b e =
( s, ( empty <<< e ||| -- a.
left <<< b % s ||| -- b.
right <<< s % b ||| -- c.
pair <<< b % s % b ||| -- d.
split <<< s' % s' ... h) -- e. ... f.
) where s' = transToN s
{-# INLINE gNussinov #-}“aPairmax” is an algebra. Algebras tell the grammar how to optimize. The only complicated thing is to use “foldl1” from Stream.Monadic in the vector package, instead of a list-based function.
aPairmax :: (Monad m) => Signature m Int Int
aPairmax = (empty,left,right,pair,split,h) where
empty _ = 0
left b s = s
right s b = s
pair l s r = if basepair l r then 1+s else -999999
{-# INLINE [0] pair #-}
split l r = l+r
h = SM.foldl1' max
basepair l r = f l r where
f 'C' 'G' = True
f 'G' 'C' = True
f 'A' 'U' = True
f 'U' 'A' = True
f 'G' 'U' = True
f 'U' 'G' = True
f _ _ = False
{-# INLINE basepair #-}
{-# INLINE aPairmax #-}This algebra pretty-prints parses and produces a dot-bracket string with the RNA secondary structure. Basically, CCCAAAGGG forms an RNA secondary structure where the C’s and G’ pair, yielding a pretty-printed string “(((…)))”.
aPretty :: (Monad m) => Signature m String (SM.Stream m String)
aPretty = (empty,left,right,pair,split,h) where
empty _ = ""
left b s = "." P.++ s
right s b = s P.++ "."
pair l s r = "(" P.++ s P.++ ")"
split l r = l P.++ r
h = return . id
{-# INLINE aPretty #-}Given that we can calculate the optimal base-pairing with “aPairmax” and pretty-print (or backtrace) results using “aPretty”, how can we combine both algebras to get backtraces of optimal parses?
We define yet another algebra "<**" that combines two algebras. No need to manually combine algebras.
While "<**" looks rather complicated, it has a number of advantages. (i) it scales nicely. Most real grammars are very complicated and separating different logics help to keep complexity down. (ii) Using template-Haskell and the Signature it seems possible to auto-generate this algebra – and we’ll try to have this in the next major version.
type CombSignature m e b =
( () -> (e, m (SM.Stream m b))
, Char -> (e, m (SM.Stream m b)) -> (e, m (SM.Stream m b))
, (e, m (SM.Stream m b)) -> Char -> (e, m (SM.Stream m b))
, Char -> (e, m (SM.Stream m b)) -> Char -> (e, m (SM.Stream m b))
, (e, m (SM.Stream m b)) -> (e, m (SM.Stream m b)) -> (e, m (SM.Stream m b))
, SM.Stream m (e, m (SM.Stream m b)) -> m (SM.Stream m b)
)
(<**)
:: (Monad m, Eq b, Eq e)
=> Signature m e e
-> Signature m b (SM.Stream m b)
-> CombSignature m e b
(<**) f s = (empty,left,right,pair,split,h) where
(emptyF,leftF,rightF,pairF,splitF,hF) = f
(emptyS,leftS,rightS,pairS,splitS,hS) = s
empty e = (emptyF e , return $ SM.singleton (emptyS e))
left b (x,ys) = (leftF b x , ys >>= return . SM.map (\y -> leftS b y ))
right (x,ys) b = (rightF x b , ys >>= return . SM.map (\y -> rightS y b))
pair l (x,ys) r = (pairF l x r, ys >>= return . SM.map (\y -> pairS l y r))
split (x,ys) (s,ts) = (splitF x s, ys >>= \ys' -> ts >>= \ts' -> return $ SM.concatMap (\y -> SM.map (\t -> splitS y t) ts') ys')
h xs = do
hfs <- hF $ SM.map fst xs
let phfs = SM.concatMapM snd . SM.filter ((hfs==) . fst) $ xs
hS phfsIt is time to combine everything. “nussinov78” prepares the input and runs the forward phase, calling “nussinov78Fill”. Then, we get all co-optimal parses using “bt”.
nussinov78 inp = (arr ! (Z:.0:.n),bt) where
(_,Z:._:.n) = bounds arr
len = P.length inp
vinp = VU.fromList . P.map toUpper $ inp
arr = runST (nussinov78Fill $ vinp)
bt = backtrack vinp arr
{-# NOINLINE nussinov78 #-}
nussinov78Fill :: forall s . VU.Vector Char -> ST s (Arr0 DIM2 Int)
nussinov78Fill inp = do
let n = VU.length inp
t' <- fromAssocsM (Z:.0:.0) (Z:.n:.n) 0 []
let t = mtblE t'
let b = Chr inp
let e = Empty
fillTable $ gNussinov aPairmax t b e
freeze t'
{-# NOINLINE nussinov78Fill #-}Backtracking is easy! Just take the grammar again, combine “aPairmax” and “aPretty”, bind the table generated in the forward phase and you should be able to extract all co-optimal parses lazily.
backtrack (inp :: VU.Vector Char) (tbl :: PA.Arr0 DIM2 Int) = unId . SM.toList . unId $ g (0,n) where
n = VU.length inp
c = Chr inp
e = Empty
t = bttblE tbl (g :: BTfun Id String)
(_,g) = gNussinov (aPairmax <** aPretty) t c e
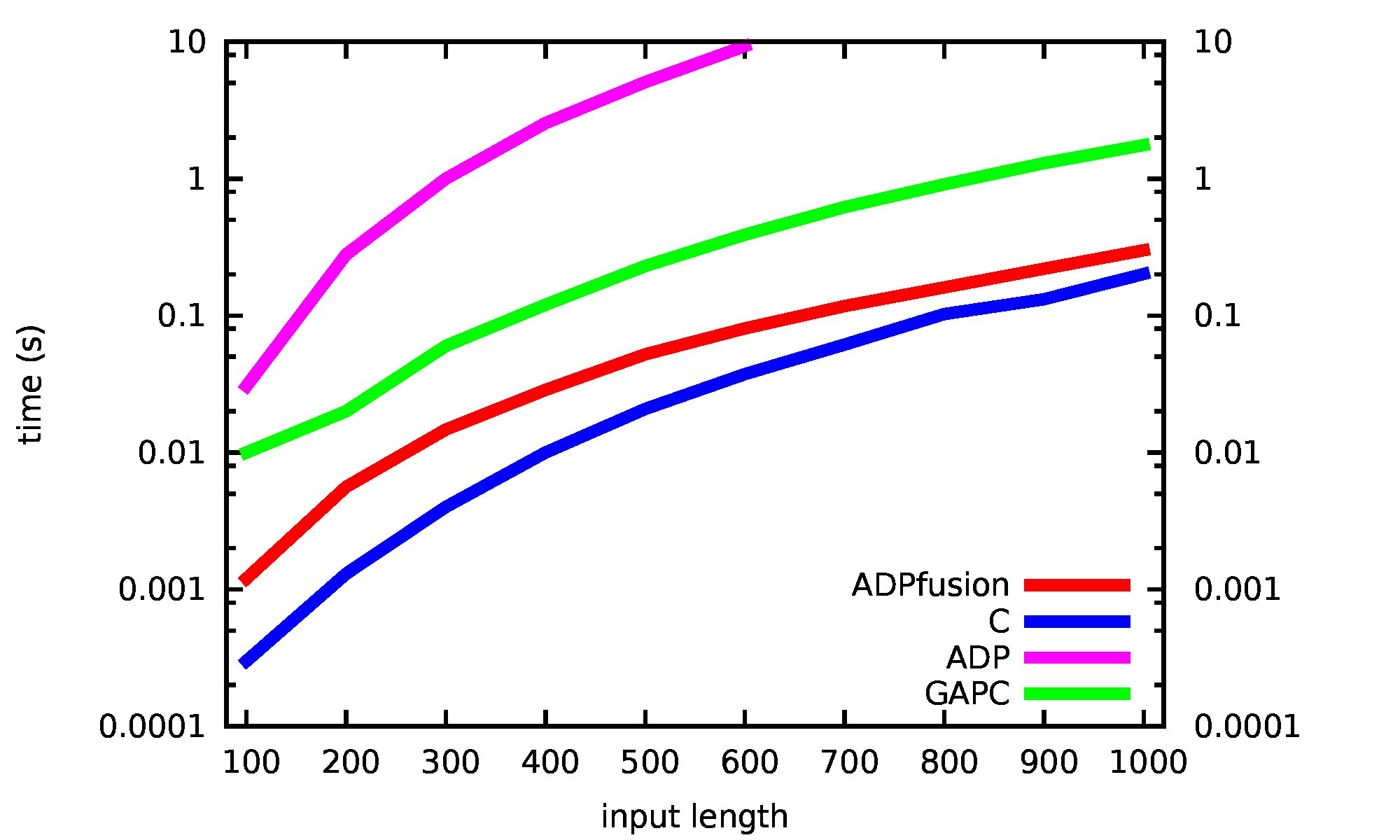
{-# INLINE backtrack #-}Runtimes: measuring up against optimized C in the real world
(all runtimes are the mean over 100 random input sequences for each 100nt step – of course, each algorithm was tested on the same input as every other)
Nussinov 78
The runtimes for Nussinov’78 are probably as good as we can hope for, using the current backend. Since we have only one table and one O(n) per cell recursion, there are not many sharing optimizations which could have been exploited in a C implementation.
This figure was produced using the new (and current) ADPfusion system. We see two fun things: (i) ADPfusion comes very close to C-code, once the input is long enough (for very small inputs, starting a haskell-program, copying around arrays, and other things just cost too much). (ii) Other frameworks are, sometimes a lot, slower. This includes orignal ADP which tends to be around x100 slower, and C++-based GAPC which we tend to beat by a factor of x8.
RNAfold v2
The RNAfold v2 algorithm uses three interdependent tables, one additional table to create the final “external” structure and a lot of lookup tables. Above, we explain the differences in implementation. Still, performance-wise Haskell is now very close to C, and performs very favorably compared to any other algorithm, except the ViennaRNA package (Lorenz et al. 2011) (look at the computation times here). We should be roughly on par with Unafold (Markham and Zuker 2008) and massively better than RNAstructure (Reuter and Mathews 2010) (about x20-x30).
All three packages aim to implement roughly the same algorithm, with some changes and have similar RNA secondary structure prediction capabilities.
The ADPfusion benchmark for RNAfold is still based on the pre-ICFP code.

paper
The pdf can be found on our preprint server.
local list of references
Ronny Lorenz, Stephan H. Bernhart, Christian Höner zu Siederdissen, Hakim Tafer, Christoph Flamm, Peter F. Stadler, and Ivo L. Hofacker. 2011. ViennaRNA package 2.0. Algorithms for Molecular Biology 6, no. 26. 6. doi:10.1186/1748-7188-6-26.
N. R. Markham, and M. Zuker. 2008. Software for nucleic acid folding and hybridization. Methods Mol. Biol 453. 453: 3–31.
Jessica S. Reuter, and David H. Mathews. 2010. RNAstructure: Software for rna secondary structure prediction and analysis. BMC bioinformatics 11, no. 1. 11: 129.