bbq tool bbq is a command-line tool for discovering clusters
of transcription factor binding sites that occur simultaneously in
several genomes. Finding such clusters – sometimes also referred
to as cis-regulatory modules – is done in a
multiple-alignment-like fashion by solving a certain combinatorial and
geometric optimization problem, the so-called best barbeque problem
(explaining the name bbq). As opposed to classical
(typically dynamic programming based) alignment procedures, the order
of the binding sites' occurences can be arbitrarily shuffled, so that
bbq is the result of developing completely new
algorithms.
The implementation provided here supports many practically
relevant features such as weighted sets, p-value based weighting
schemes or computing the best h solutions instead of only
the best solution, to mention the most important ones. Most of these
features are supported by two algorithms representing two different
approaches to solving the given optimization problem.
Suppose we are in the following situation:
We have a set of 6 short DNA fragments that are potential binding sites for transcription factors. Furthermore, we are given a set of 3 genome sequences, GS1, GS2 and GS3. We suspect that a subset of the 6 binding sites occurs within a cluster of some limited length – say 50 nucleotides – on each of the 3 genomes. Our goal is to decide whether there is a reasonably large subset of fragments occuring clustered in each genome sequence: if we find such clusters containing a significant number of fragments, it can be suspected that the concerted effect of these binding sites fullfils a certain task and hence has been evolutionary conserved.
Our first assumption (which is simplified and to be refined later) is that clusters containing a large number of binding sites are more likely to be functionally relevant than clusters containing a small number of fragments. Hence, we want to maximize the number of elements in the cluster.
In order to solve this problem using bbq, we set up a
file containing a description of the 6 potential binding sites:
c414M2 GGCTGCGAA 1
|
We save this file as bsites.motifs.
Each line describes one binding site. The first column specifies a name and the second column the corresponding DNA sequence, while the third column specifies a number of admissible mismatches. In the example above, this means that occurences of fragments with 1 mismatching character are also taken into account.
Now we want to plug this information into bbq, together
with the three genomes and our suspected cluster length 50. We do this by
typing the command line
bbq 50 bsites.motifs GS1.fa GS2.fa GS3.fa
|
c106M1,
c196M3 and c196M4. Since the rest of the
output looks rather machine- than human readable, we apply option
-S to obtain some more details:
bbq -S 50 bsites.motifs GS1.fa GS2.fa GS3.fa
|
We obtain the following output:
|
This gives us all information on occurences of the individual
fragments within the clusters. Note that, for instance, the cluster in
GS3.fa contains c106M1 in addition to the
three fragments {c106M1,c196M3,c196M4}, which is not
found in the other two genome sequences' clusters.
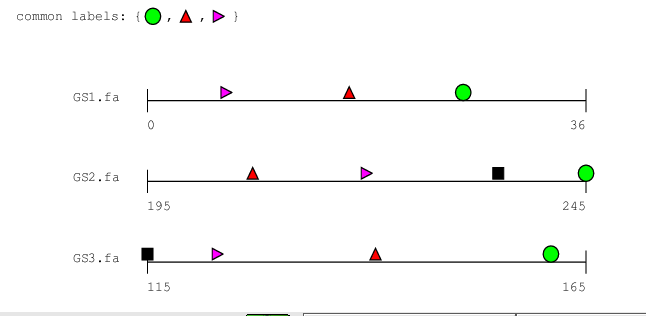
Graphical Output
Although option -S gives us all details on the occurences
within the clusters, the textual output is somewhat hard to read, and
we would be much happier with a graphical
representation. bbq supports graphical output in
postscript format as shown in the following example:
bbq -S -P bsites.ps 50 bsites.motifs GS1.fa GS2.fa GS3.fa
|
This command produces a graphical representation of the clusters found
by bbq. Using your favourite postscript viewer, you will
see something like this in bsites.ps:
Which shapes correspond to which fragments is written to a another
postscript file called labels.ps.
>Reverse Occurences
In the instances described so far, bbq only takes into
account binding site occurences on the 5' strand. In many cases,
however, we want to take into account occurences on the 3' strand as
well. We can tell bbq to do so by specifying option -r:
bbq -r -P bsites-r.ps 50 bsites.motifs GS1.fa GS2.fa GS3.fa
|
Looking at the output, we see that taking into account reverse matches yields a better solution involving four binding sites:
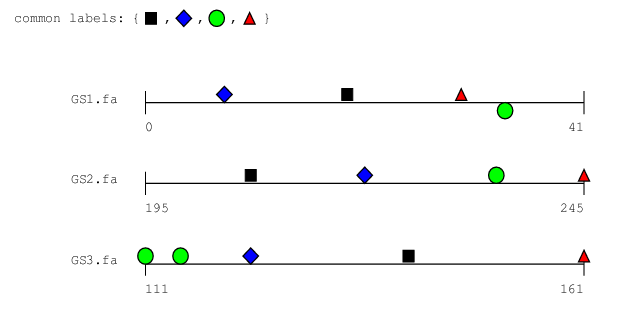
Binding sites occuring on the 5' strand are printed on top of the line indicating the DNA strand, while occurences on the 3' strand are printed below this line.
Note that binding site c106M1 (indicated by a green
circle) occurs on the 3' strand in both GS1.fa and
GS2.fa, while in GS3.fa, there only is an
occurence on the 5' strand.
bbq Command Synopsis
As we have seen in the examples above, the bbq command
uses the following command line format:
bbq [options] <length> <motif-file> <seq-file1> ... <seq-fileK>
|
where <length> is an integer specifiying the
maximum length for the clusters to be detected,
<motif-file> is a file containing the TFBS
sequences along with their names and mismatch thresholds, while
<seq-file1> ... <seq-fileK> are
Fasta-formatted files containing the sequences to be aligned. Note
that in the current version, each sequence file may only contain one
single seuqence.
The options we have encountered so far are -S for
more detailled output, -r for taking into account both
strands and -P <ps-filename> for producing
postscript output. Further options will be discussed in the sequel.
Grouping and Applying Weighting Schemes
Grouping
In some instances, we might encounter different alternatives for
one binding site that bind to the same transcription factor. In this
case, we want occurences of two (or more) binding sites to be treated
as the occurence of one binding site type. To this end,
bbq allows us to group several binding sites
together. This is done by considering prefixes of equal length. As an
example, consider the case that we want to treat the three fragments
whose name starts with c196 as one group:
c196M2 AAGTAATTAGT 1
|
We simply do this by passing a prefix length of 4 to
bbq, sucht that all sequence names whose prefix of length
4 is equal will be grouped together. This is achieved by passing
-g 4 as an option to bbq. Now,
bbq -g 4 -S -P bsites.ps 50 bsites.motifs GS1.fa GS2.fa GS3.fa
|
yields the best clusters of binding site groups.
Weighting Schemes
A somewhat critical assumption in the considerations so far was to
assume that maximizing the number of binding sites simultaneously
occuring in one cluster determines the "most interesting"
solution. However, it is easy to construct instances involving many
very short fragments along with few very long fragments. It appears to
be straightforward that clusters containing some of the few long
sequences should be ranked much higher than clusters containing the
same or even a larger number of short fragments. In this situation,
bbq supports weighting schemes, so that instead of
finding largest cardinality clusters, we rather search for maximum
weighted clusters. Two different weighting schemes are supported by
specifying option -wp or -wm. Option
-wp yields a weighting scheme based on p-value-like
measures for fragment occurences (based on dinucleotide distributions
of the underlying sequences), while -wm takes into
account the number of mismatches of occurences. It is highly
encouraged to use -wp, since -wm exists for
rather historical reasons.
Applying -u tells bbq to use the
unweighted, cardinality-based scoring scheme for clusters, which also
is the default setting. In addition to weighting schemes, one can
activate the use of multisets using option
-m. Using multisets, multiple occurences of the same
fragment within a cluster will be weighted in their sum. Without using
multisets, only the best weighted fragment of each type will be taken
into account. In some cases, multisets might give trouble with
Algorithm (A2) (see below).
Getting more than One Solution
So far, we used bbq only to find the best solution,
either invloving the largest number of binding sites or the best
weighted set of binding sites. However, it might happen that the
second-, third- or tenth-best solution rather than the best ranked
solution contains the desired conserved structure. Furthermore, we
typically want to know whether the score of the best solution is
significantly better than the score of other solutions – which would
point up the significance of the best solution.
Two options allow for finding non-optimal solutions: -t
<threshold> allows to define a threshold value, so that
bbq outputs all solutions whose score is at least
<threshold>, while -h
<num_of_hits> computes the
<num_of_hits> best ranked solutions. Used in combination with
-P, bbq produces one postscript file for each solution. For instance,
bbq -h 3 -S -P bsites.ps 50 bsites.motifs GS1.fa GS2.fa GS3.fa
|
produces output files bsites0.ps,
bsites1.ps and bsites2.ps.
For both of these options there are some caveats that we briefly
discuss next.
Using Thresholds
Typically, -t yields
most, but not all solutions exceeding the given
threshold. In most cases, in particular for small thresholds, there
will be an abundant number of solutions. Before producing output, we
might want to know how many solutions there are and, if there are too
many solutions, raise the threshold until we obtain a reasonable
number of solutions. In fact, this can be achieved using
bbq's -tc <threshold> option:
bbq -tc 2.5 -wp -S -P bsites.ps 50 bsites.motifs GS1.fa GS2.fa GS3.fa
|
tells us – without producing any further output – that there are 280 solutions whose p-value-based score is at least 2.5. Since we do not want to inspect 280 solutions by hand, we run
bbq -tc 25.5 -wp -S -P bsites.ps 50 bsites.motifs GS1.fa GS2.fa GS3.fa
|
telling us that there are five solutions whose p-value-based score is at least 25.5, so that we can easily inspect the five postscript files by hand after running
bbq -t 25.5 -wp -S -P bsites.ps 50 bsites.motifs GS1.fa GS2.fa GS3.fa
|
Finally, note that the number of thresholding solutions obtained by
the two different algorithms implemented in bbq may
differ significantly. On choosing among the two algorithms, we refer
to Section Further Options.
Computing the Best h Solutions
Using option -h seems to be straightforward – just
tell bbq how many solutions you want and then get the
best possible solutions. Unfortunately, things are not quite that
simple. The reason for complications is that solutions whose score is
close to the optimal score tend to be very similar to the best
soltion. As a user, however, we usually want to see solutions that
look significantly different from the optimal solution. To this end,
bbq usues certain heuristics to find "significantly
different" solutions. However, these heuristics might fail to yield
different solutions. Future versions of bbq might provide
better solutions.
Further Options
Choosing Algorithms
As mentioned before, bbq supports implementations of
two different algorithms. Which one is used can be specified by
-A1 (deafult) or -A2. Algorithm
(A2) will typically run faster than (A1) when more than two genome
files are involved, while (A1) should be used when only two genomes
are involved. In fact, the running time of (A2) can often be observed
to decrease with the number of genome seuqences involved! Finally,
note that -A2 may yield suboptimal solutions in
conjunction with -m.
Reporting Progress
On larger instances, running bbq might take very long
– in fact, the optimization problem that bbq deals with
is NP-hard. In order to get an idea how much time remains, you can
specify option -p in order to get a steady report on what
part of the overall search space that bbq examines has
been visited yet. While usually working reliably in conjunction with
Algorithm (A1), progress reports may run very unsteady in conjunction
with (A2).
References
[Abstract]
Detecting Phylogenetic Footprint Clusters by Optimizing Barbeques
Axel Mosig,
Türker Biyikoglu,
Sonja J. Prohaska,
Peter F. Stadler
Submitted, 2004