
| Sequence ID | dm3.chr2L |
|---|---|
| Location | 8,869,593 – 8,869,650 |
| Length | 57 |
| Max. P | 0.960626 |
| Location | 8,869,593 – 8,869,650 |
|---|---|
| Length | 57 |
| Sequences | 4 |
| Columns | 57 |
| Reading direction | forward |
| Mean pairwise identity | 57.44 |
| Shannon entropy | 0.66417 |
| G+C content | 0.29308 |
| Mean single sequence MFE | -6.17 |
| Consensus MFE | -3.29 |
| Energy contribution | -5.10 |
| Covariance contribution | 1.81 |
| Combinations/Pair | 1.67 |
| Mean z-score | -1.12 |
| Structure conservation index | 0.53 |
| Background model | dinucleotide |
| Decision model | sequence based alignment quality |
| SVM decision value | 1.53 |
| SVM RNA-class probability | 0.947562 |
| Prediction | RNA |
| WARNING | Out of training range. z-scores are NOT reliable. |
Download alignment: ClustalW | MAF
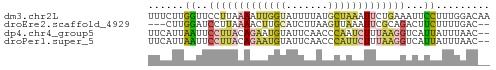
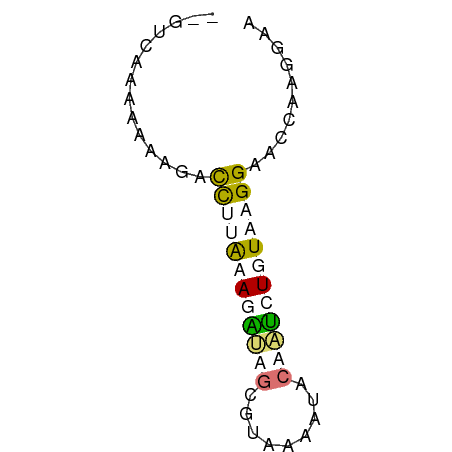
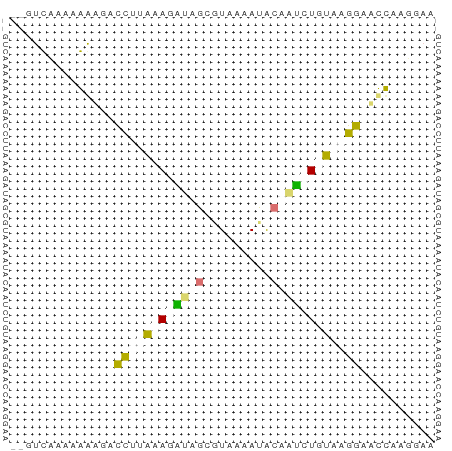
>dm3.chr2L 8869593 57 + 23011544 UUUCUUGGUUCCUUAAAAUUGGUAUUUUAUGCUAAAUUCUGAAAUUCCUUUGGACAA ....(((((....((((((....)))))).)))))..((..((.....))..))... ( -5.80, z-score = 0.22, R) >droEre2.scaffold_4929 9462998 52 + 26641161 ---CUUGGAUCCUUAAAACUUGCAUCUUAAGUUAAAUUCGCAGACUUCUUUUGAC-- ---.............((((((.....))))))......................-- ( -2.20, z-score = 1.46, R) >dp4.chr4_group5 1676968 55 - 2436548 UUCAUUAAUUCCUUACAGAAUGUAUUCAACCCAAUCUUUAAGGUCAUUAUUUAAC-- .....((((.(((((.(((.((.........)).))).)))))..))))......-- ( -6.50, z-score = -1.94, R) >droPer1.super_5 1648074 55 - 6813705 UUCAUUAAUUCCUUACAGAAUGUAUUCAACCCAUUCUUUAAGGUCAUUAUUUAAC-- .....((((.(((((.((((((.........)))))).)))))..))))......-- ( -10.20, z-score = -4.24, R) >consensus UUCAUUAAUUCCUUAAAAAAUGUAUUCAACCCAAAAUUCAAAGACAUCAUUUAAC__ ......((..(((((((((((((.......)))))))))))))...))......... ( -3.29 = -5.10 + 1.81)
| Location | 8,869,593 – 8,869,650 |
|---|---|
| Length | 57 |
| Sequences | 4 |
| Columns | 57 |
| Reading direction | reverse |
| Mean pairwise identity | 57.44 |
| Shannon entropy | 0.66417 |
| G+C content | 0.29308 |
| Mean single sequence MFE | -7.83 |
| Consensus MFE | -4.53 |
| Energy contribution | -6.03 |
| Covariance contribution | 1.50 |
| Combinations/Pair | 1.50 |
| Mean z-score | -1.01 |
| Structure conservation index | 0.58 |
| Background model | dinucleotide |
| Decision model | sequence based alignment quality |
| SVM decision value | 1.68 |
| SVM RNA-class probability | 0.960626 |
| Prediction | RNA |
| WARNING | Out of training range. z-scores are NOT reliable. |
Download alignment: ClustalW | MAF
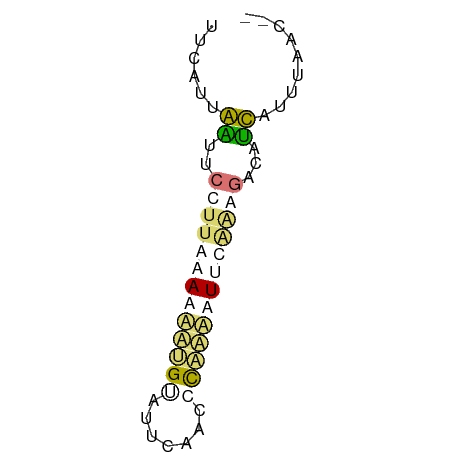
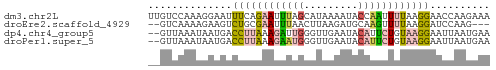
>dm3.chr2L 8869593 57 - 23011544 UUGUCCAAAGGAAUUUCAGAAUUUAGCAUAAAAUACCAAUUUUAAGGAACCAAGAAA ...(((....(((((....)))))....((((((....)))))).)))......... ( -3.80, z-score = 0.60, R) >droEre2.scaffold_4929 9462998 52 - 26641161 --GUCAAAAGAAGUCUGCGAAUUUAACUUAAGAUGCAAGUUUUAAGGAUCCAAG--- --........................((((((((....))))))))........--- ( -5.50, z-score = 0.35, R) >dp4.chr4_group5 1676968 55 + 2436548 --GUUAAAUAAUGACCUUAAAGAUUGGGUUGAAUACAUUCUGUAAGGAAUUAAUGAA --......((((..(((((.(((.((.........)).))).))))).))))..... ( -9.40, z-score = -1.61, R) >droPer1.super_5 1648074 55 + 6813705 --GUUAAAUAAUGACCUUAAAGAAUGGGUUGAAUACAUUCUGUAAGGAAUUAAUGAA --......((((..(((((.((((((.........)))))).))))).))))..... ( -12.60, z-score = -3.38, R) >consensus __GUCAAAAAAAGACCUUAAAGAUAGCGUAAAAUACAAUCUGUAAGGAACCAAGGAA ..............((((((((((((.........)))))))))))).......... ( -4.53 = -6.03 + 1.50)
Generated by rnazCluster.pl (part of RNAz 1.0) on Tue Apr 19 21:27:11 2011